Синдром Сотоса или синдром церебрального гигантизма – врожденное, обычно спорадическое (единичное, проявляющееся нерегулярно) заболевание, которое характеризуется высокорослостью, черепно-лицевой дисморфией, костными изменениями и разной степенью умственной отсталости (обычно наблюдается умеренная степень).
| МКБ-10 | Q87.3 |
|---|---|
| МКБ-9 | 759.89 |
| DiseasesDB | 29134 |
| OMIM | 117550 |
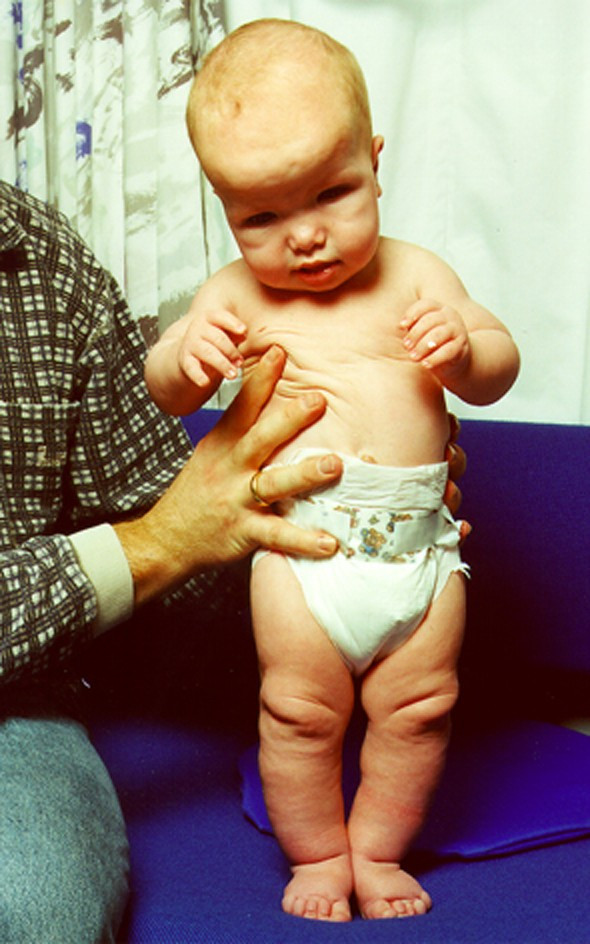
Люди с данным синдромом отличаются выступающими лобными буграми, грубыми чертами лица, увеличенным расстоянием между парными органами (гипертелоризм), косоглазием, антимонголоидным разрезом глаз, выступающей вперед нижней челюстью, высоким небом и аномально большим языком (макроглоссия). Кисти и стопы большие, возможно полное или частичное сращение пальцев стоп. Характерен также кифосколиоз.
Костный возраст превышает паспортный, а эндокринный статус не отклоняется от нормы.
Общие сведения
Синдром Сотоса впервые описал в 1964 г.американский педиатр Хуан Фернандес Сотос. До настоящего времени было описано около 120 случаев наличия данной патологии, поэтому ее частота к настоящему времени не установлена.
Отмеченный спорадический характер большинства известных случаев дал основания отнести синдром Сотоса к аутосомно-рецессивным заболеваниям, но в последнее время установлен аутосомно-доминантный тип наследования, а нерегулярный характер заболевания связывают с:
- этиологической неоднородностью генетических мутаций;
- частотой проявления наличия патологического гена;
- гонадным мозаицизмом, вызванным мутациями в половых клетках на ранней стадии их развития при отсутствии мутаций в соматических клетках родителей.
Синдром чаще встречается у мальчиков.
Формы
Выделяется два типа синдрома Сотоса:
- Тип 1 вызывается мутацией гена NSD1 при существовании двух альтернативных аллелей (гетерозиготное состояние) или при хромосомных перестройках с утратой части хромосомы в регионе 5q35. Кодируемый данным геном белок является белком-корегулятором, который влияет на активность андрогенных рецепторов, регулирующих процесс транскрипции. Заболевание носит аутосомно-доминантный характер, но в большинстве случаев развивается благодаря мутациям, происходящим в момент зачатия (мутация “de novo”).
- Тип 2 вызывается мутацией гена NFIX. Данный ген принимает участие в регуляции транскрипции, взаимодействуя с эстрогеновыми рецепторами.
Причины развития
Синдром церебрального гигантизма возникает в результате мутаций гена NSD1 (5-я хромосома) или гена NFIX. Согласно недавно появившейся гипотезе, локализация гена синдрома Сотоса – короткое плечо 3-ей хромосомы, регион р21.
Мутация может происходить благодаря аутосомно-доминантному типу наследования, при котором мутантный вариант наблюдается хотя бы у одного из родителей. Дефектный аллель доминирует над нормальным вариантом, поэтому проявляется как при гомозиготном (в результате слияния идентичных половых клеток (гамет)), так и при гетерозиготном (в результате слияния несколько отличающихся вариантов половых клеток) состоянии.
При аутосомно-доминантном типе наследования патологию можно обнаружить, изучив родословную “по вертикали”.
Чаще причиной заболевания служат мутации “de novo”, которые не встречаются у родителей, но затрагивают половые клетки и происходят у эмбрионов после их оплодотворения.
Симптомы
Церебральный гигантизм манифестирует с момента рождения. Обычно дети с данным синдромом рождаются крупными и по сравнению со средними данными отличаются значительной длиной (около 55 см) и массой (от 3900 кг) тела.
Осмотр новорожденного не всегда позволяет сразу выявить наличие синдрома, но практически всегда выявляется:
- антимонголоидный разрез глаз;
- выступающие лобные бугры;
- высокое готическое небо;
- прогнатизм (увеличенная в разной степени нижняя челюсть);
- макроглоссия, при которой из-за большого размера языка возможны дыхательные нарушения и трудности сосания;
- желтуха новорожденных.
Также выявляются выраженные в разной степени:
- Макродолихоцефалия (увеличение размеров головы без избыточного скопления в желудочках мозга цереброспинальной жидкости).
- Гипертелоризм. Обычно наблюдается глазной гипертелоризм, при котором увеличено расстояние между внутренними уголками глаз и зрачками.
- Страбизм (косоглазие) любого типа.
- Увеличенные стопы и кисти (акромегалия). Может наблюдаться вальгусная стопа и полное или частичное сращение пальцев стопы (синдактилия).
- Кифосколиоз, при котором позвоночник искривлен одновременно в сагиттальной (проходящая спереди назад воображаемая ось делит тело на левую и правую часть) и фронтальной плоскости.
Синдром Сотоса у детей может сопровождаться:
- Висцеромегалией, при которой обычно наблюдается увеличенный объем почек, сердца или печени.
- Пороками развития сердца. Чаще всего наблюдается дефект межпредсердной перегородки и дисфункция клапанов, возможна атрезия легочной артерии, межпредсердное сообщение, нарушения ритма и проводимости сердца и др.
- Пороками развития почек или центральной нервной системы.
- Судорогами и нарушением координации.
- Вегетативными дисфункциями (повышенная потливость, недостаточный контроль температуры).
Дети с синдромом Сотоса интенсивно растут, особенно в первые 3-4 года жизни. Психомоторное развитие происходит с некоторой задержкой – при неосложненном родовой травмой и наличием тяжелых пороков заболевании держать голову дети начинают около 3-х месяцев, сидеть – приблизительно в 7-м месяцев, а ходить – около 1,5 лет и позже. Походка у таких детей неустойчивая.
Мимическая реакция развивается также с задержкой, обычно нет реакции на игрушки.
Зубы прорезываются рано.
Речь развивается поздно – у большинства больных детей отдельные слова появляются к 2-м годам при наличии импрессивной речи (понимания речи окружающих). Запас слов у разговаривающих детей ограничен (в отдельных случаях при способности произносить все звуки речь не формируется даже к 2,5 годам).
Эмоциональная сфера и поведение нарушены – дети обычно малоактивны, замкнуты, легко возбуждаются, не контролируют эмоции и склонны проявлять агрессию.
Часто отмечается недоразвитость большой моторики и плохая координация малых движений кистей рук.
Прием пищи часто затруднен.
При умеренной умственной отсталости дети могут ориентироваться в окружающей обстановке и иметь навыки самообслуживания. Обучаемость сохранена.
Диагностика
Диагностика основывается на данных осмотра и типичной для заболевания симптоматике.
Критериями служит наличие:
- высоких показателей длины и массы тела при рождении;
- ускоренного роста;
- макродолихоцефалии;
- акромегалии;
- нарушений координации;
- умеренной умственной отсталости.
Для постановки диагноза применяют:
- рентгенофункциональные методы, которые позволяют выявить несоответствие костного возраста паспортному;
- ЭКГ, которая показывает наличие неспецифических изменений;
- ЭЭГ, которое может выявить нарушение взаимоотношений коры и подкорковых образований;
- ЭХО-ЭГ, позволяющее выявить наличие умеренного расширения боковых желудочков мозга;
- МРТ, которая позволяет иногда выявить гипоплазию мозолистого тела.
Определяется также кариотип больного, который при синдроме Сотоса соответствует норме.
Диагноз подтверждается путем дифференциальной диагностики и данных генетического анализа.
Подтверждение диагноза требует определения:
- уровня соматотропного гормона (СТГ) для исключения гигантизма, вызванного чрезмерной выработкой гормона роста;
- костного возраста;
- содержания и спектра аминокислот в сыворотке и моче для исключения гомоцистинурии;
- уровня 17-гидроксипрогестерона, чтобы исключить врожденную гиперплазию коры надпочечников;
- уровня глюкозы для исключения синдрома Беквита—Видемана;
- уровня эстрогенов или андрогенов для исключения преждевременного полового развития;
- данных цитогенетического анализа, чтобы исключить синдром Клайнфельтера.
Отграничить синдром от болезни Марфана помогает клиническая картина и определение гармоничности телосложения больного – гармоничным физическим развитием отличаются только больные с синдромом Сотоса.
Лечение
Специфического лечения для синдрома Сотоса не существует. Больным назначаются:
- Симптоматическая терапия.
- Ноотропы, стимулирующие умственную деятельность и улучшающие способность к обучению. Обычно назначают длительные курсы энцефабола или аминалона.
- Витамины.
- Сосудистые средства.
Рекомендуются также физиопроцедуры, занятия с логопедом, остеопатом, а также курсы реабилитации в центрах, занимающихся детьми с наличием подобных патологий.
Профилактика
Поскольку синдром Сотоса – генетическое заболевание, в настоящее время профилактика не разработана.