Болезнь Краббе (галактозилцерамидный липидоз или глобоидно-клеточная лейкодистрофия) – редко встречающееся наследственное заболевание, которое относится к группе лизосомных болезней накопления (вызываются нарушением функции лизосом). Проявляется в поражении миелиновой оболочки нервных волокон, прогрессирующей мозговой дегенерации, повышении мышечного тонуса, очень высокой температуре тела (гиперпирексии) и умственной отсталости.
| МКБ-10 | E75.2 |
|---|---|
| МКБ-9 | 330.0 |
| DiseasesDB | 29468 |
| MeSH | D007965 |
| OMIM | 245200 |
| eMedicine | ped/2892 |
Общие сведения
Первое описание заболевания относится к 1916 году и принадлежит датскому неврологу Кнуду Гаралденсену Краббе, поэтому заболевание названо в его честь.
Распространенность заболевания – 1 случай на 100 000 человек. Более часто болезнь Краббе встречается на Скандинавском полуострове – 1 к 50000, а также у живущих на израильской территории арабов – 1 к 6000.
Болезнь встречается у представителей обоих полов в равной степени.
Это заболевание может также наблюдаться у кошек и собак (преимущественно у мелких пород терьеров).
Формы
В зависимости от возраста, в котором проявилось заболевание, выделяют следующие клинические формы галактозилцерамидного липидоза:
- инфантильную или классическую (развитие болезни начинается с 3-6 месяцев);
- позднюю инфантильную (с 6-18 месяцев);
- ювенильную;
- взрослую.
Классическая форма составляет 85-90 % всех случаев.
Причины развития
Болезнь Краббе вызывается мутациями в GALC гене, который расположен на 14 хромосоме в регионе q31.
Ген GALC обеспечивает синтез фермента галактоцерамидазы (галактозилцерамид-b-галактозидазы). Этот фермент расщепляет простейший гликолипид галактоцереброзид на галактозу и церамид.
Галактоцереброзид является важным компонентом миелина, который образует защитное покрытие вокруг нервных волокон, обеспечивающих быструю передачу нервных импульсов.
При обусловленной мутацией гена GALC недостаточности фермента в головном мозге и других органах происходит отложение аномально большого количества нерасщепленной производной галактоцереброзида – психозина (галактозилсфингозина). Значительное накопление психозина токсично для клеток, формирующих миелиновую оболочку, поэтому она постепенно разрушается. В результате процесс дегенерации затрагивает не только ЦНС, но и периферические нервы.
Болезнь наследуется по аутосомно-рецессивному типу.
Патогенез
При недостаточности фермента галактоцереброзидазы или белка сапозина А, который необходим для «распознавания» ферментом субстрата, нерасщепленные гликолипиды и их производная психозин накапливаются в мозге, почках, селезенке, печени, лейкоцитах и клетках соединительной ткани (фибробластах). В результате количество психозина превышает норму в 10-100 раз.
Для нервной системы психозин токсичен, поскольку он вызывает гибель клеток нейроглии (олигодендроцитов), которые обеспечивают миелинизацию аксонов. В зонах распада миелиновой оболочки в нервной ткани вокруг кровеносных сосудов образуются характерные включения – глобоидные гистиоциты (являются макрофагами, которые способны захватывать и переваривать бактерии и т.д.).
Гибель олигодендроцитов сопровождается повреждением нейронов, которые являются основной структурно-функциональной единицей мозга. Место отмерших нейронов заполняется клетками нейроглии и развивается глиоз.
Аксональная дегенерация затрагивает и периферические нервы, в которых накапливаются пенистые гистиоциты.
Болезнь быстро прогрессирует.
Симптомы
Симптомы болезни Краббе зависят от формы заболевания.
Выделяют три стадии инфантильной формы болезни (развивается до 6 месяца).
Для I стадии характерно наличие неспецифических симптомов, которые проявляются в:
- повышении мышечного тонуса (спастический тип);
- гипервозбудимости;
- немотивированных подъемах температуры;
- трудностях при вскармливании.
На 6-м – 8-м месяце жизни проявляются нарушения психомоторного развития, возможны судороги.
II стадия заболевания сопровождается:
- быстрой утратой приобретенных ранее навыков;
- внезапными хаотическими сокращениями отдельных мышц (миоклония);
- нарастанием мышечного тонуса до судорожной позы, при которой спина резко выгнута дугой и опора идет только на пятки и затылок (опистотонус);
- снижением интеллекта;
- атрофией зрительных нервов, при которой снижается реакция зрачков на свет;
- снижением или отсутствием сухожильных рефлексов;
- гипотрофией вплоть до крайнего истощения (кахексия).
Для III стадии характерны:
- судороги;
- развитие бульбарно-псевдобульбарного синдрома, при котором нарушается процесс глотания и произношения звуков, а также наблюдается потеря звучности голоса;
- утрата функций головного мозга.
Симптомами поздней инфантильной формы являются:
- рано развивающееся поражение зрительного или нерва сетчатки, которое при отсутствии недостатков органов зрения приводит к слепоте (может быть частичной или полной);
- постепенное снижение интеллекта;
- нарушения двигательных навыков.
К симптомам ювенильной и взрослой формы относятся:
- Зрительная агнозия (неспособность узнавать и классифицировать поступающую зрительную информацию) или гемианопсия, при которой выпадает половина зрительного поля.
- Спастические парезы и параличи, которые возникают благодаря прогрессирующей мозжечковой атаксии и невропатии. Выявляются в первую очередь при нарушениях походки.
Диагностика
Диагностика болезни Крабе включает:
- Изучение жалоб и анамнеза, при котором уточняется, в каком возрасте появились первые симптомы, как быстро развивается болезнь и т.д.
- Изучение семейного анамнеза, при котором уточняется, наблюдались ли подобные симптомы болезни у родственников.
- Общий осмотр. В процессе осмотра оценивается мышечный тонус, выраженность сухожильных рефлексов, координация движений, походка и др.
- Анализ спинно-мозговой жидкости (ликвора), при котором оценивается прозрачность и цвет жидкости, ее давление, количество глюкозы, белка (повышается в процессе разрушения нейронов), солей хлора.
- Биохимические тесты, которые заключаются в измерении уровня фермента галактоцерамидазы или в обнаружении повышенного уровня психозина. Для анализа берется кровь или клетки кожи.
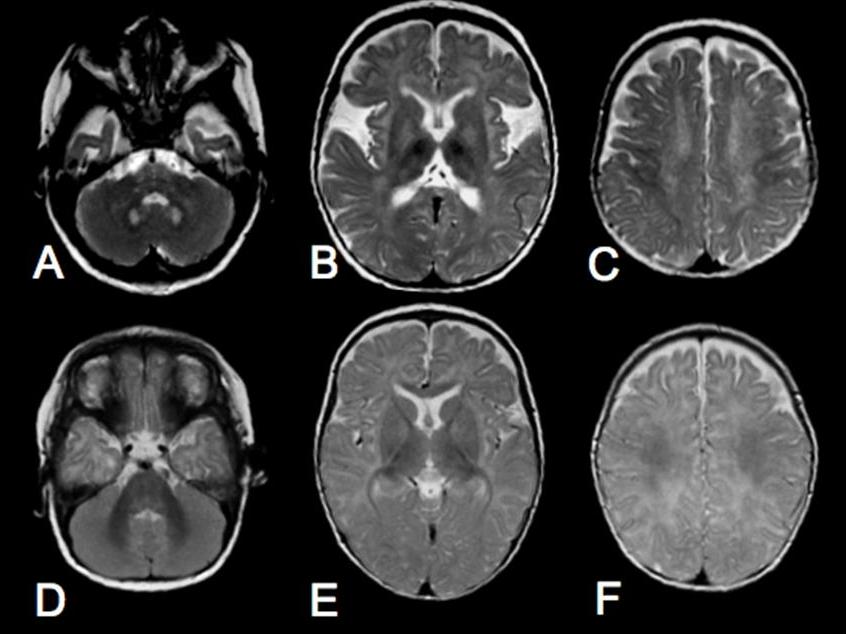
- КТ, МРТ и ЭНМГ (электронейромиография). На начальной стадии заболевания при помощи МРТ выявляется поражение подкорковых структур, белого вещества мозжечка и пирамидальных трактов. Для поздних стадий характерна атрофия большого мозга, поражение задних отделов ствола мозга или валика, поражение белого вещества головного мозга в теменно-затылочных отделах. ЭНМГ позволяет обнаружить пониженную скорость проведения импульса по периферическим нервам, а также их демиелинизацию.
- Молекулярно-генетическое обследование.
Болезнь Краббе допускает пренатальную диагностику, в процессе которой исследуется амниотическая жидкость. В исследуемых амниотических клетках определяется активность галактоцерамидазы.
Лечение
Эффективное лечение болезни Краббе в настоящее время находится в стадии разработки – ученые изучают возможность доставки Galc гена в клетки больного при помощи вирусов (генная терапия), однако этот метод до конца не изучен.
На ранних стадиях заболевания или при медленно прогрессирующих формах достаточно эффективным методом лечения является трансплантация:
- костного мозга, которая позволяет стабилизировать состояние больного и снизить проявления симптомов;
- пуповинной крови, которая останавливает дальнейшее развитие заболевания.
Кроме того, проводится симптоматическое лечение, включающее прием противосудорожных препаратов и др.
Прогноз неблагоприятный – при инфантильной форме заболевания летальный исход наступает на протяжении 2-х лет. При поздних формах заболевания и медленном прогрессировании болезни продолжительность жизни увеличивается.
Профилактика
Болезнь Краббе относится к наследственным заболеваниям, поэтому единственной возможной профилактикой является генетический анализ при наличии болезни Краббе у близких родственников.
Болезнь проявляется при наличии дефектного гена у обоих родителей (вероятность 1 к 4).
При наличии мутации гена GALC у одного из родителей человек является просто носителем дефектного гена.