Лиссэнцефалия (в переводе с др.-греч. «гладкий головной мозг») – аномалия развития, для которой характерно сглаживание извилин коры больших полушарий головного мозга.
| МКБ-10 | Q04.3 |
|---|---|
| МКБ-9 | 742.2 |
| OMIM | 236670 247200, 253280, 253800, 257320, 300121, 300215, 304800, 601545, 611603 |
| DiseasesDB | 29492 |
| MeSH | D054082 |
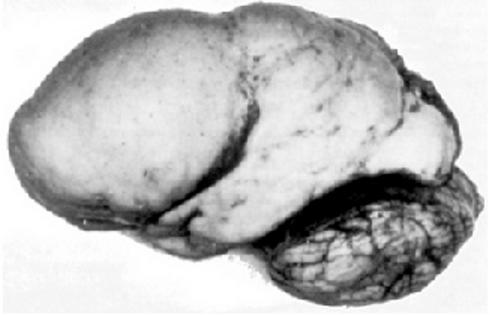
Возникает вследствие недостаточной миграции зародышевых нервных клеток (нейробластов) из первичной нервной трубки.
Наиболее тяжелая форма заболевания – агирия, при которой извилины мозга вообще отсутствуют.
Общие сведения
Лиссэнцефалия – редко встречающийся порок развития, который может быть как самостоятельной аномалией развития, так и проявлением некоторых врожденных синдромов (Миллера-Дикера и Нормана-Робертса). Наблюдается у детей независимо от пола. Распространенность заболевания в настоящее время не установлена.
Формы
Лиссэнцефалия может быть тотальной и очаговой.
В зависимости от тяжести нарушений выделяют:
- агирию, при которой поверхность мозга лишена извилин;
- пахигирию, которая отличается наличием нескольких плоских широких извилин и неглубоких борозд.
В зависимости от морфологических и генетических данных с 2003 года выделяется пять групп данной аномалии.
Первая группа – классический тип заболевания (I тип лиссэнцефалии в предшествующей классификации), которая включает:
- лиссэнцефалию, связанную с мутацией гена LIS1;
- лиссэнцефалию, которая вызывается мутацией гена DCX (doublecortin);
- изолированную лиссэнцефалию I типа;
- синдром Миллера-Дикера;
- изолированную лиссэнцефалию I типа, которая возникла по неустановленным генетическим причинам.
Во вторую группу включена форма, которая связана с X-сцепленным рецессивным наследованием и сопровождается врожденным отсутствием мозолистого тела. Заболевание вызывается мутацией гена ARX.
Третья группа включает лиссэнцефалию, при которой наблюдается врожденное недоразвитие (гипоплазия мозжечка). К этой же группе относится синдром Норман-Робертс, вызванный мутацией гена RELN.
К четвертой группе относят микролиссэнцефалию.
В пятую группу входит «булыжниковая» лиссэнцефалия (II тип по старой классификации). Включает:
- синдром Уокера-Варбурга (HARD(E)-синдром);
- синдром Фукуямы;
- Мышечно-Глазо-Мозговую Болезнь (MEB).
Существует также морфологическая классификация, выделяющая:
- 1 степень, которая ставится при полной диффузной агирии без контурирующихся извилин.
- 2 степень, которая ставится при наличии «расширенной» агирии в сочетании с присутствием отдельных извилин (обычно эти извилины расположены в височных и лобных отделах).
- 3 степень – при обширных участках агирии и пахигирии. В большинстве случаев пахигирия отмечается в лобных отделах, а агирия – в теменных и центральных отделах мозга
- 4 степень ставится при «расширенной» пахигирии (участки агирии отсутствуют).
- 5 степень ставится при наличии симметричных участков пахигирии, которые перемежаются с фрагментами коры, соответствующей норме.
Причины развития
Лиссэнцефалия развивается в результате нарушения миграции нейронов при формировании и развитии коры головного мозга.
Нарушение миграции возможно в результате мутации:
- гена RELN (рилина), который расположен на хромосоме 7 в регионе 7q22 (развивается синдром Норман-Робертс);
- гена ARX, расположенного на Х-хромосоме в регионе p21.3.
К нарушениям формирования коры головного мозга приводит и частичная утрата (делеция) гена LIS-1, расположенного в хромосоме 17 регион р13.3 и принимающего участие в миграции нейронов (аутосомно-рецессивный тип наследования).
При утрате некоторых генов 17-й хромосомы в регионе 13 (PAFAH1B1) развивается синдром Миллера-Дикера.
К развитию лиссэнцефалии приводят также нарушения X-сцепленных генов. При мутациях гена XLIS у детей мужского пола диагностируют лиссэнцефалию, а у детей женского пола – двойную кору, так как в результате нарушения миграции слой серого вещества откладывается под белым веществом головного мозга (в норме должно быть наоборот). Дефектный ген кодируется белком doublecortin (DCX), поэтому его мутация тоже может стать причиной развития аномалии. Тип наследования – Х-сцепленный рецессивный.
Причиной развития «булыжниковой» лиссэнцефалии являются:
- Дефекты 9-й хромосомы в регионе q34 (синдром Уокера-Варбурга).
- Дефекты гена FCMD, расположенного в 9-й хромосоме в регионе q31 (синдром Фукуямы). Чаще всего мутация заключается в присутствии избыточных мобильных генетических элементов первого типа.
- Мутации гена POMGNT1, который расположен в 1-й хромосоме в регионе p34 (Мышечно-Глазо-Мозговая Болезнь).
Причины некоторых форм лиссэнцефалии в настоящее время не выявлены.
Кроме генетических нарушений причиной развития аномалии может быть:
- перенесенная в первом триместре вирусная инфекция матки или плода;
- плохое кровоснабжение мозга плода на начальном этапе беременности.
Патогенез
Формирование и развитие коры головного мозга включает:
- пролиферацию (деление) нервных клеток;
- миграцию нейронов из эмбрионального матрикса;
- дифференцировку коры.
При нормальном развитии плода происходит:
- оформление полушарий мозга на 11-12 неделе внутриутробного развития;
- формирование латеральной, шпорной и опоясывающей борозды на 16 неделе эмбриогенеза;
- уплощение кортикального плато и формирование первичных и центральных борозд на 20 неделе;
- формирование ольфакторных борозд на 24-26 неделе;
- образование верхних височных борозд на 28 неделе.
Созревание коры головного мозга обеспечивается миграцией нейронов, которые располагаются в мозге слоями. В процессе миграции те клетки, которые образовались позже, мигрируют дальше своих предшественниц, образуя таким образом новый слой.
Процесс миграции регулируется белками рилином и N-кадгерином.
Большая часть борозд и извилин формируются после окончания миграции нейронов, поэтому нарушение процесса миграции нейронов приводит к нарушению формирования извилин и борозд.
Симптомы
Симптомы заболевания зависят от типа лиссэнцефалии.
Классический тип заболевания проявляется:
- отставанием в росте, психоречевом и двигательном развитии;
- относительно небольшим размером головы;
- судорогами, которые могут манифестировать с первых дней жизни или развиваться после 1,5 лет;
- симптомокомплексом «вялого ребенка».
Лиссэнцефалия при синдроме Миллера-Дикера сопровождается:
- черепно-лицевой дизморфией (изменением формы лица);
- увеличенным расстоянием между парными органами;
- замедленным ростом;
- патологиями почек, сердца, ЖКТ.
При обследовании выявляется уменьшенное количество кортикальных слоев (четыре вместо шести).
При синдроме Нормана-Робертса заболевание сопровождается:
- увеличенным расстоянием между глазами;
- микроцефалией (маленький размер головы);
- наличием покатого лба;
- эпилепсией;
- отеком тканей (лимфедемой);
- задержкой умственного развития.
Обследование позволяет выявить утолщенную кору мозга, наличие агирии, значительные изменения гиппокампа, мозжечка и ствола мозга.
При синдроме Уокера-Варбурга помимо лиссэнцефалии наблюдается:
- микрофтальмия, катаракта, отслоение сетчатки;
- аномалии развития скелета и мышц;
- гидроцефалия, цефалоцеле;
- задержка умственного развития.
При синдроме Фукуямы лиссэнцефалия сочетается с:
- прогрессирующей мышечной дистрофией;
- задержкой темпов развития;
- выраженным слабоумием;
- гидроцефалией, микрополигирией, аплазией мозолистого тела, уменьшением плотности белого вещества;
- судорогами в большинстве случаев.
В отдельных случаях выявляют атрофию зрительных нервов и патологию сетчатки.
Диагностика
Для диагностики лиссэнцефалии используются:
- пренатальное обследование на 22- 28 неделе беременности при помощи ультразвукового оборудования;
- КТ, позволяющая выявить наличие сглаженности извилин, гипоплазию белого вещества, увеличение желудочков мозга, прямолинейную границу между серым и белым веществом, расширение и вертикальную ориентацию сильвиевых щелей, расширение центральной борозды и др.;
- МРТ, которая позволяет выявить тип лиссэнцефалии по характерным признакам;
- ЭЭГ, при помощи которого определяется характерный для лиссэнцефалии быстрый и высокоамплитудный ритм в задних отделах мозга;
- генетический анализ.
При пренатальной диагностике необходимо учитывать, что в начале II-го триместра достаточно гладкий мозг – норма, поэтому оценивать формирование извилин и борозд можно не раньше 20 недели.
При подозрении на наличие аномалии возможно исследование хромосом плода.
Лечение
Специфическое лечение лиссэнцефалии не разработано.
Для улучшения состояния больных применяются:
- производные вальпроевой кислоты в качестве противосудорожной терапии;
- карнитин, который используется при мышечной дистрофии;
- церебрализин, актовегин, глицин, витамины в качестве поддерживающей терапии.
В некоторых случаях возможно оперативное вмешательство, которое заключается в трансплантации стволовых клеток. Поскольку положительные результаты пока наблюдаются редко и заключаются только в некотором улучшении состояния, а риск побочных эффектов у больного ребенка высок, операции проводят не часто.
Лиссэнцефалия – заболевание с неблагоприятным прогнозом, поэтому семьям, в которых имелось данное заболевание, необходима консультация генетика.